JavaScript seems to be disabled in your browser. For the best experience on our site, be sure to turn on Javascript in your browser.
Tel: +1-832-696-8203
Email: [email protected]
Worldwide Distributors
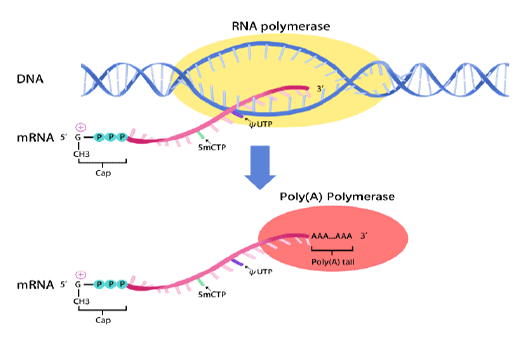
In vitro transcription of capped mRNA with modified nucleotides and Poly(A) tail
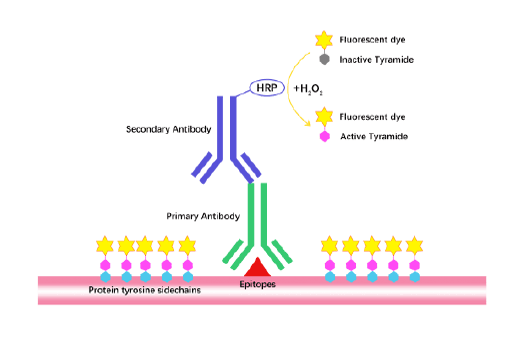
TSA (Tyramide Signal Amplification), used for signal amplification of ISH, IHC and IC etc.
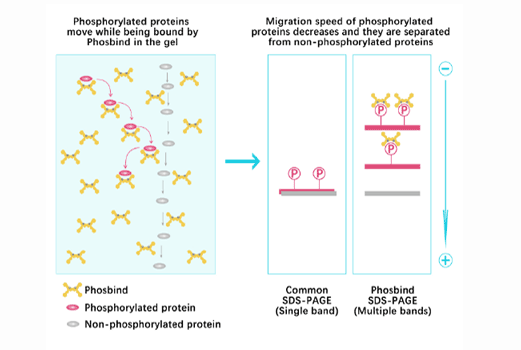
Separation of phosphorylated and non-phosphorylated proteins without phospho-specific antibody
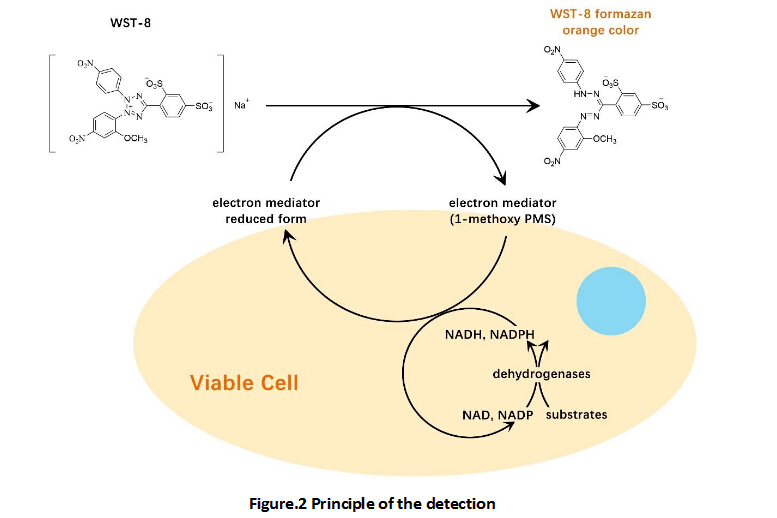
A convenient and sensitive way for cell proliferation assay and cytotoxicity assay
Protect the integrity of proteins from multiple proteases and phosphatases for different applications.
Valspodar is a potent inhibitor of P-glycoprotein (P-gp) widely used in preclinical and clinical studies [1].P-gp is a transmembrane glycoprotein which is located on cell membrane. P-gp distributes extensively and is expressed in certain cell types primarily containing liver, colon, kidney and pancreas. It also is known as multidrug resistance protein 1 (MDR1) which is pumps foreign substances out of cells. P-gp decreases the net uptake of cytotoxic drugs into the cells and mediats the efflux of these agents out of the cells, which is ATP-dependent. P-gp also overexpress in some cancer cells. P-gp plays an important role in mediating resistance to anticancer drugs and decreasing drug accumulation in multidrug-resistant cancer cells.[1] Valspodar can reverse the resistance to mitoxantrane which is due to the expression of P-gp. The IC50 of mitoxantrane decreased from 1.6 ± 0.13 μM to 0.4 ± 0.02μM in MDA-MB-435mdr cells pretreated with 3 mg/ml PSC. Valspodar increase the mitoxantrane intracellular accumulation by decreasing drug efflux and increasing mitoxantrone net uptake in cells.[1] The cytotoxicity was significant greater in T47D/TAMR-6 cells treated with doxorubicin and valspodar than doxorubicin only. Co-encapsulation of doxorubicin and valspodar presents a promising anticancer effect.[2] Valspodar was rapid absorpted and reachs the peak within 2 hnafter an oral dose. Valspodar showed properties of wide distribution, low hepatic extraction and mean bioavailability of 42.8% in rat.[3] References: [1]. Shen F, Bailey BJ, Chu S, Bence AK, Xue X, Erickson P, Safa AR, Beck WT, Erickson LC: Dynamic assessment of mitoxantrone resistance and modulation of multidrug resistance by valspodar (PSC833) in multidrug resistance human cancer cells. J Pharmacol Exp Ther 2009, 330(2):423-429.[2]. Bajelan E, Haeri A, Vali AM, Ostad SN, Dadashzadeh S: Co-delivery of doxorubicin and PSC 833 (Valspodar) by stealth nanoliposomes for efficient overcoming of multidrug resistance. J Pharm Pharm Sci 2012, 15(4):568-582.[3]. Binkhathlan Z, Hamdy DA, Brocks DR, Lavasanifar A: Pharmacokinetics of PSC 833 (valspodar) in its Cremophor EL formulation in rat. Xenobiotica 2010, 40(1):55-61.
Cell lines
MDR-P388 murine leukemia cells
Reaction Conditions
26 ~ 70 μM valspodar
Applications
Valspodar restored sensitivity of MDR-P388 murine leukemia cells to cytostatic agents when used at concentrations of 70, 33, 45, 34, and 26 nM in combination with colchicine, vincristine, daunomycin, doxorubicin, and etoposide, respectively.
Animal models
MDR-P388 tumor-bearing B6D2FI mice
Dosage form
25 and 50 mg/kg
Administered orally 4h before each doxorubicin treatment
The combined therapy resulted in a marked valspodar dose-dependent prolongation of the mean survival time (MST) of the MDR-P388 tumor-bearing mice. At the doses of 25 and 50 mg/kg, valspodar could extent the MST to 28.7 and 38.8 days, respectively, when administered 4 h before each doxorubicin intraperitoneal injection. Thus, valspodar could serve as a very promising chemosensitizer.
Note
The technical data provided above is for reference only.
References:
1. Boesch D, Gavériaux C, Jachez B, et al. In vivo circumvention of P-glycoprotein-mediated multidrug resistance of tumor cells with SDZ PSC 833. Cancer Research, 1991, 51(16): 4226-4233.